什么是MDR临床评估报告?
临床评估报告(CER)是MDR中的一份重要的技术文件。有效的临床评估报告描述了对所有可用临床证据的结构化评估和分析,以此评估医疗器械的安全性和性能。
虽然临床评估是一个持续的过程,但临床评估报告按照设备的风险分类间隔提供临床评估结论的阶段性结论。无论风险等级如何,制造商都必须针对EU MDR下的所有医疗器械生成临床评估报告。
由于结构合理的临床评估报告是MDR下所有医疗器械监管批准的关键要求,因此制造商必须了解新立法下开发CER的要求。和欧杰这类专业的MDR咨询机构,无疑是制造商达成目标的一个捷径。
根据MDR撰写临床评估报告的要求是什么?
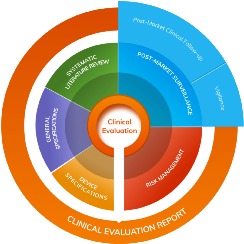
生成临床评估报告(CER)所需数据的活动包括系统文献综述,上市后监测、上市后临床随访、警惕和风险管理
图1:生成临床评估报告(CER)所需数据的活动
MDR第61条规定,每个医疗器械的临床评估应记录在CER中,CER必须构成该设备技术文档组合的一部分。MDR附件XIV A部分扩展了这一要求,并提供了进行临床评估的详细要求,要求该过程符合以下要求:
基于记录在案的临床评估计划
识别与设备相关的所有临床证据,无论是否有利
根据规定的方案以稳健的方式评估和分析临床数据
制定计划以解决临床证据组合中的任何差距
就医疗器械的安全性和性能得出结论
虽然附件XIV A部分概述了如何进行临床评估,但对编写CER的要求相对较轻。它仅规定“临床评估的结果应记录在临床评估报告中,该报告应支持对设备符合性的评估。
关于编写CER的更详细指南可在 MedDev 2.7/1 rev 4“临床评估:制造商和公告机构的指南”中找到,这是欧盟委员会发布的一份咨询文件。它提供了关于CER结构的有用部分,但该指南尚未更新(截至2022年9月),以反映MDR引入的要求的变化。因此,仅遵守 MedDev 2.7/1 rev 4 还不足以实现 MDR 合规性,制造商需要使用其他信息和专业知识来源,以确保满足欧盟 MDR 要求。
临床评估报告应如何构建?
根据 MedDev 2.7/1 修订版 4,医疗器械临床评估报告应至少包含以下信息:
医疗器械详情
对设备所在的临床领域的审查,包括该领域的替代疗法和对现有技术的评估
评估与设备及其临床性能相关的临床数据
上市后监测(PMS)、警戒系统和上市后临床随访(PMCF)活动的结果
效益风险评估
关于设备是否符合相关的MDR附件一般安全和性能要求(GSPR)并适合其预期用途的客观结论
上面包含的每个部分都应仔细构建和精确编写,以确保可能不具备医疗器械专业知识的评估人员可以理解该文件,这点确实没办法,我们都想当然认为欧盟的TD评估人员应该是非常专业、非常了解咱们的产品,事实上他们可能对法条比我们要了解,但是对于具体的产品,他们会非常的外行。
前面我们讲到了自MedDev 2.7 /1 Rev 4发布以来,还没有根据MDR的规定进行更新,因此另一份文件医疗器械协调小组(MDCG)发布的MDCG 2020-13 就显得非常非常的重要,该文件设定了MDR评估医疗器械临床评估报告的标准。提出了一些具体的要求,包括:
要求分析与可比较替代设备相关的临床证据,以确定可量化的安全性和性能基准,从而可以据此评估受试者设备。
包括CER中的临床评估计划摘要。
在可能的情况下,对CER中考虑的临床证据进行统计分析。
医疗器械等效性的使用限制。
详细说明如何在收益风险分析中证明临床益处,包括需要量化和基于证据的所谓益处。
根据欧盟MDR,临床评估报告应对临床证据进行有力、客观、科学的分析。CER编写者使用的语言必须传达对设备安全性和性能的平衡评估,该评估必须足够强大,以满足监管审查的要求。CER最好由具有足够程度的医学、科学和监管经验的人编写,以满足这些要求。
临床评估报告中的临床证据有什么具体要求?
临床证据的鉴定、评估和分析是根据MDR编写CER的核心要求。临床证据以下两个方面被认为是CER的两个关键组成部分:
1:与可比较的替代设备相关联,以便建立安全和性能基准,以便与受试者设备进行比较
评估受试者设备的安全性和性能时
2:必须确定所有相关的临床证据,无论是有利的还是不利的,并且必须根据临床评估计划中指定的方案进行评估和分析。还必须有避免重复数据的具体程序。
与受试者设备相关的临床证据来自两个来源 - 由制造商生成和持有,以及独立研究人员产生并在文献中发表的证据。
制造商生成的证据包括从上市前研究、PMS、警戒和 PMCF 活动中生成的证据。
独立证据必须通过一个强有力的、有案可稽的搜索协议来识别,该协议将包含确保找到所有相关证据的规定。
证据评估涉及考虑以下因素:
方法和样本量的适用性
与受试者设备的相关性及其临床评估
研究类型和质量
潜在的偏倚
所采用的统计分析技术的适用性
在识别和评估之后,临床评估报告应包含对临床证据的分析,以确定受试者设备是否履行了MDR下的安全性和性能义务(包括与可比较的替代设备进行比较)。
有效撰写临床评估报告需要获得临床证据识别、评估和分析方面的高级专业知识,欧杰公司在此方面具有十七年的临床评估报告咨询经验,我们的参与可以让制造商在CER方面走更少的弯路,节约大量的时间和费用,把专业的问题留个专业的老师解决,永远是最正确的事情。
CER完成后会该怎么办?
临床评估报告完成后,CER将作为TD文件的重要组成部分,提交公告机构进行审核评估。进行CER评估的程序取决于设备的风险分类。
根据MDR, IIa类,IIb类和III类医疗器械的制造商必须将完成的CER提交给公告机构(或英国脱欧后英国的英国批准机构)进行监管审查,作为合格评定程序的组成部分。除非临床评估报告符合MDR要求,否则不会授予加盖CE标志的许可。MDR概述了公告机构在对临床评估报告进行评估方面的责任,要求公告机构具有足够医疗科学和技术专业知识的个人,以便对CER进行合理的评估。临床评估报告评估将遵循MDCG 2020-13中规定的指导方针。
I类装置的制造商通常不需要向公告机构提交CER进行评估。但是,I类设备仍需遵守 MDR 要求才能进行临床评估,因为它必须证明符合附件I GSPR、是否适合预期目的以及可接受的益处风险状况。因此,I类设备仍然需要临床评估报告,并且应该具有与高风险设备所需的类似程度的谨慎和技能。
此外,某些类别的I类设备有特殊要求,可能需要由公告机构进行评估。提供无菌(Is类)、可重复使用的手术设备或具有测量功能(Im类)的I类设备将需要公告机构的参与,这些类型的设备的制造商在准备CER时应考虑到公告机构的参与。
在哪里可以得到撰写临床评估报告的帮助?
对于根据MDR寻求批准的器械,CER必须达到足够的标准,这一点极为重要。强烈建议聘请对欧盟MDR合规性要求有全面了解的经验丰富的CER作者,比如我们欧杰的专业MDR顾问老师。利用专业帮助可能是一项明智的投资,因为不符合所需监管标准的CER可能会一次又一次的被公告机构退回,每一次退回都会导致认证时间的拖延和认证费用的增加,事实上很多客户两到三年都没有拿到CE证书,基本上都是卡在了CER报告这一环节。
使用临床评估报告模板可能是开始起草CER的有效方法,我们欧杰公众号的下载单元有现成的CER模板,有需要的网友可以自己去免费下载,但必须注意能否有效利用现成模板还取决于使用者的医学专业知识以及识别、评估和分析临床数据的能力。